CellR4 2014; 2 (4): e1077
Rapidly progressive renal and hepatic failure in AL-amyloidosis: bortezomib and steroid support in a young woman two months after delivery
Topic: Stem cells
Category: Case Reports
Abstract
MATERIALS AND METHODS: We describe a 41 years old woman admitted to the hospital with proteinuria (1280 mg/24h) and rapid deterioration of renal function (serum creatinine from 0.8 to 1.6 mg/dL). Autoantibodies, immunoglobulin and C3/C4 were negative. A renal biopsy showed the presence of AL-Amyloidosis with k-light chains deposition at immunofluorence. Subsequently, the patient showed nephrotic syndrome onset (proteinuria 4000 mg/24h with albuminuria 3400 mg/24h) and increased rates of cholestasis with hepatomegaly and hepatic failure.
RESULTS: Treatment with bortezomib and dexamethasone gave a complete hematological response but renal function was not improved.
DISCUSSION: This case is very interesting because renal involvement was the initial presentation of amyloidosis and rapid progressive renal and hepatic failure was subsequentely observed; its management was challenging from the clinical approach to the final diagnosis and treatment.
CONCLUSIONS: In terms of organ response, it is necessary to develop new strategies to counteract the progressive organ failure due to amyloid deposition.
ABBREVIATIONS: AL-amyloidosis, light chain amyloidosis; GFR, glomerular filtration rate; HDM, high dose melphalan; SCT, stem cell transplantation; sCr, serum creatinine; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, Gamma-glutamyl transferase; AP, alcaline phosphatase; INR, International Normalized Ratio; BNP, B-type natriuretic peptide; HDM/SCT, high-dose melphalan followed by autologous stem cell transplantation.
INTRODUCTION
Amyloidosis is a rare disease with higher prevalence in adult males; its diagnosis is difficult because of the absence of early manifestations and specific signs. Disease includes a heterogeneous group of pathological features characterized by the extracellular deposition of fibrillar proteinaceous material in various tissues and organs in particular renal glomeruli with an estimated prevalence ranging from 2.7% to 4% of renal biopsies 1. The marked selective proteinuria (albuminuria), usually reported, reflects the main glomerular deposition that characterizes this kind of damage. Amyloidosis is defined light chain amyloidosis (AL) when the protein fibrils derived from the variable region of the light chain, usually lambda chains deposits 2 , 3. The most common manifestations of AL-amyloidosis are related to the involvement of kidney, heart and liver 4. The symptoms depend on the organs affected by the accumulation of amyloid.
The goal of treatment is eradication of the monoclonal plasma cell population and suppression of the pathologic light chains, which can result in organ improvement and extend patient survival. Standard treatment approaches include oral melphalan and prednisone, even if only a quarter of patients achieved a hematologic response to this treatment and the median survival was only 12-18 months 5. In fact a progressive glomerular filtration rate (GFR) reduction with cardiac involvement and increased in liver size usually occurred despite therapy. In this context, high dose melphalan (HDM) followed by autologous hematopoietic stem cell transplantation (SCT) has shown to be effective in reducing clonal disease and circulating light chains improving kidney function in AL-amiloidosis nephropathy 6 and is not associated to the numerous side effects related to chemotherapy. Recently, therapeutic strategies may include novel agents such as immunomodulatory drugs or proteasome inhibitors, in particular bortezomib, in patients not eligible for SCT.
MATERIALS AND METHODS
We have studied a 41 years old woman admitted to the Department of Clinical Medicine, Nephrology and Dialysis Unit, Policlinico Umberto I of Rome, in December 2011 due to recent onset of proteinuria and rapid deterioration of renal function (serum creatinine – sCr from 0.8 to 1.6 mg/dL during the last 2 months). She had a child 2 months before admission and during that period she didn’t present proteinuria but only high blood pressure. She did not refer nephrotoxic drugs assumption but frequent episodes of epistaxis. On admission in our Nephrology Department she was apyretic and hydrated in appearance without any peripheral edema, she had mild heart murmur with blood pressure 150/85 mmHg and heart rate 75 bpm; the rest of physical examination was negative. Laboratory tests showed anemia with haemoglobin 10 g/dL, red blood cells 3560 x 103/mm3 and serum iron 100 mg/dL, hypercholesterolemia and hypertriglyceridemia with cholesterol level 260 mg/dL and triglyceride level 300 mg/dL, white blood cells 5680/mm3, platelets 230 x 103/mm3, glucose 84 mg/dL, blood urea nitrogen (BUN) 14.5 mg/dL, uric acid 5 mg/dL, serum sodium 140 mEq/L, serum potassium 4.3 mEq/L, serum calcium 9.5 mg/dL, serum phosphate 4 mg/dL, serum protein 6.5 g/dL, aspartate aminotransferase (AST) 15 U/L, alanine aminotransferase (ALT) 17 U/L, coagulation test and serum protidogram were normal. Blood gas analysis was normal. Urinalysis: pH 6.5, specific gravity 1015, red blood cells absent. The 24 h urine volume was 2 liters with proteinuria 1280 mg/24 h. Renal ultrasonography excluded ureteral obstruction and showed kidney normal size. We suspected an autoimmune disease but ANA, dsDNA, c/p-ANCA, ENA antibodies were negative and IgG/A/M, C3, C4 were in the normal range. Furthermore TSH, FT3, FT4, markers of hepatitis B and C viruses resulted in normal range. We performed a renal biopsy which showed global glomerular deposition of pale eosinophilic acellular weakly PAS-positive material deposited in the mesangial region and along capillaries, obliteration of capillary lumen and patent bowmans’ space (Figure 1); this material was Red Congo positive (Figure 2) and immunofluorescence shows conspicuous mesangial accumulations of AL amyloid that stain with anti-Kappa light chains antiserum (Figure 3).
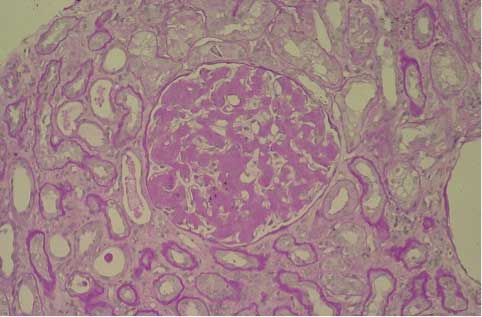
Figure 1. Extensive glomerular deposition of amorphous material. PAS 200x

Figure 2. The glomerulus stain with Congo red. 200x

Figure 3. Immunofluorescence shows conspicuous mesangial accumulations of AL amyloid that stain with anti-Kappa light chains antiserum.
RESULTS
Two months after delivery, our patient presented a rapid deterioration of renal function with nephrotic syndrome and deterioration of liver function associated to hepatomegaly: sCr 2.0 mg/dl, BUN 40.5 mg/dL, albumin 3.0 mg/dl, total bilirubin 1.9 mg/dL, AST 200 U/I, ALT 150 U/I, Gamma-glutamyl transferase (GGT) 50 U/I, alcaline phosphatase (AP) 25 U/I, International Normalized Ratio (INR) 1.5, 24h urine test showed proteinuria 4000 mg/24 h with albuminuria 3400 mg/24 h and immunoelectrophoresis showed abnormal zone of restriction in kappa light chain, suggesting Bence-Jones protein, free kappa type (Figure 4); hepatic ultrasound showed diffuse coarse echo and hypoechoic pattern with hepatomegaly; renal ultrasound showed normal kidney size with increase of parenchymal echogenicity, mildly reduced cortical thickness and resistance indices increased bilaterally. At the same time, cardiac biomarkers increased showing a possible extent of cardiac involvement, even if echocardiogram was negative: troponin T 30 ng/L (normal values in woman 0-14 ng/L) provided a quantitative assessment of cardiac damage, B-type natriuretic peptide (BNP) and NT-proBNP resulted increased and indicated cardiomyocyte stress 7.

Figure 4. A-C, Progression of renal and liver impairment during 4-5 months: renal function was evaluated with serum creatinine (mg/dl), proteinuria and microalbuminuria (g/24 hours); hepatic function was evaluated with AST, ALT, GGT and AP (U/L).
AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, Gamma-glutamyl transferase; AP, alcaline phosphatase.
The patient was treated with bortezomib 1.3 mg/m2 biweekly associated with dexamethasone. The treatment led to a mild clinical improvement and rapid hematological response (1 month) with good tolerance of therapy. Complete hematological response was defined on the basis on the International Society for Amyloidosis criteria as negative serum and urine immunofixation electrophoresis, normal serum free light chain ratio and < 5% clonal plasma cells on bone marrow studies 8. However, patient showed a sustained worsening of renal function with a progressive GFR reduction until 20 ml/min; therefore she has been referred to a predyalisis satellite unit.
DISCUSSION
The epidemiology of AL-amyloidosis is not well known because it is a rare entity difficult to recognize, due to the unspecific early clinical manifestations of the disease. The hypothesis of AL-amyloidosis is only considered when specific organ failure occurs. Clinical/organ involvement were defined by the consensus report from the 10th International Symposium on Amyloid and Amyloidosis as demonstration by biopsy of affected organ or biopsy at an alternate site to confirm the histologic diagnosis of amyloidosis: fine-needle abdominal fat aspirate and/or biopsy of the minor salivary glands, rectum, or gingiva 9. In our case the unusual nephrologic onset consisting of hypertension and nephrotic proteinuria in young woman after pregnancy imposed the renal biopsy that finally defined the diagnosis of AL-amyloidosis. Renal biopsy demonstrated the amyloid glomerular deposition consists of kappa light chains, while lambda light chains are observed more frequently. The marked selective proteinuria, mainly represented by albumin (about 80%), expresses the main glomerular damage typical of fibrillar amyloid deposition 10. Moreover a rapid progressive renal and hepatic failure was simultaneously observed, and renal amyloid deposition was restricted to the glomeruli, whereas myocardium was not involved initially.
Subsequently cardiac involvement occurred despite therapy and cardiac biomarkers were increased. In fact, several studies suggest that Troponin I or T provide a quantitative assessment of cardiac damage and are independently associated with survival 11. Moreover B-type natriuretic peptide (BNP) and/or NT-proBNP can be useful to recognize cardiomyocyte stress earlier than cardiac ultrasonography. Patients with cardiac involvement have a mean survival of 1.1 years after diagnosis, and a survival less than 6 months if treatment is not provided once the first symptoms of hearth failure are recognized 12 , 13 , 14 especially if the signs of heart failure persist when the diagnosis is confirmed.
The occurrence of disease after pregnancy demonstrates its protective effect as reported in the literature 15. In fact clinical and experimental evidence suggests that female sex hormones can modulate the inflammatory response. Progesterone has a strong anti-inflammatory effect that is mediated by several mechanisms, including inhibition of the transcription nuclear factor kappa-B, enhancement of corticosteroid production and induction of kallikrein binding protein synthesis by the liver. Estrogens were also shown to exert anti-inflammatory effects by inhibition of T cell-induced inflammation suppression of polymorphonuclear leukocytes, suppression of serum amyloid component and induction of kallikrein binding protein synthesis by the liver.
The goal of treatment is the eradication of the monoclonal plasma cell population and suppression of the pathologic light chains that can result in organ improvement and extend patient survival. This clone of plasma cells is the source of the amyloidogenic light chains, histologically identical to those seen in the more common plasma cell dyscrasia, multiple myeloma. Given these similarities, treatments for AL-amyloidosis have been largely derived from those studied for the treatment of multiple myeloma 16 , 17. The efficacy of a treatment can be measured both in terms of reduction in the burden of clonal plasma cell disease (hematologic response) and by improvement in the organ function (organ response) 18. The first effective treatment for AL-amyloidosis was HDM and prednisone. However, only a quarter of patients achieved a hematologic response to this treatment and the median survival was only 12-18 months 19. High-dose melphalan followed by autologous stem cell transplantation (HDM/SCT) was explored in AL-amyloidosis based on its success in treating multiple myeloma. Two large studies from experienced centers confirmed the utility of HDM/SCT as a treatment for AL-amyloidosis. At Boston University, 312 patients with AL amyloidosis were treated with HDM/SCT at 200 mg/m2 or 140 mg/m2 based on age and cardiac status. Utilizing a multidisciplinary team for peri-transplant management, mortality related to treatment was reduced to 14% in these selected patients 20. In this series, the median survival for those who achieved complete response was more than 10 years compared to 50 months for those who did not achieve complete response 21. A second large patient series from the Mayo Clinic reported 434 patients with AL-amyloidosis treated with HDM/SCT over 14 years. A hematologic response was seen in 76% of patients including 39% who achieved complete response. Treatment related mortality was 10% 22.
Despite the reported efficacy, the use of HDM/SCT in AL-amyloidosis remains controversial. In fact only 20-25% of patients with AL-amyloidosis are eligible for such aggressive treatment 23. Strategies for those not eligible for transplantation include novel agents such as proteasome inhibitors. Targeting the proteasome, the cellular machinery largely responsible for protein homeostasis was rational based on the misfolded nature of proteins in AL-amyloidosis. In our case the patient was treated with dexamethasone and bortezomib, a reversible inhibitor of the 26S proteasome, with a complete hematological response and good tolerance of the treatment 24. However, in terms of organ response our patient showed a sustained and rapid worsening of renal function despite therapy and a progressive GFR reduction until 20 ml/min.
CONCLUSIONS
Treatment with bortezomib and steroids can be an effective approach for AL-amyloidosis in order to reduce clonal plasma cell and give a complete hematological response. However, new approaches have to be developed in order to also improve organ response targeting amyloid deposit and harnessing the immune system.
ACKNOWLEDGMENT: We thank Konstantinos Giannakakis who performed histopathological examination of renal biopsy and revised histopathological description.
Conflict of Interests: The Authors declare that they have no conflict of interests.
REFERENCES
- Gesualdo L, Di Palma AM, Morrone LF, Strippoli GF, Schena FP, Italian Immunopathology Group, Italian Society of Nephrology. The Italian experience of the national registry of renal biopsies. Kidney Int 2004; 66(3): 890-894. (back)
- Pinney JH, Hawkins PN. Amyloidosis. Ann Clin Biochem 2012; 49(Pt3): 229-241. (back)
- Oe Y, Nakaya I, Yahata M, Murata O, Yaegashi H, Sakuma T, Sato H, Liepnieks JJ, Benson MD, Soma J. κ I light chain AL amyloidosis presenting with rapidly progressive renal and hepatic failure with unusual renal amyloid distribution. Clin Nephrol 2012; 77(1): 66-70. (back)
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 2011; 97(1): 75-84. (back)
- Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336: 1202-1207. (back)
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011; 18: 47. (back)
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart 2011; 97(1): 75-84. (back)
- Palladini G, Dispenzieri A, Gertz MAA. Validation of the criteria of response to treatment in AL amyloidosis. Blood 2010; 116: 586-587. (back)
- Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins FN, Merlini G, Moreau P, Ronco P, Sanchorawala V, Sezer O, Solomon A, Grateau G. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. Am J Hematol 2005; 79: 319-328. (back)
- Shafique S, Wetmore J, Almehmi A. Primary amyloidosis of the kidney. W V Med J 2010; 106(1): 22-24. (back)
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011; 18: 47. (back)
- Madan S, Dispenzieri A, Lacy MQ, Buadi F, Hayman SR, Zeldenrust SR, Rajkumar SV, Gertz MA, Kumar SK. Clinical features and treatment response of light chain (AL) amyloidosis diagnosed in patients with previous diagnosis of multiple myeloma. Mayo Clin Proc 2010; 85(3): 232-238. (back)
- Sedaghat D, Zakir RM, Choe J, Klapholz M, Saric M. Cardiac amyloidosis in patient with multiple myeloma: A case report and review of literature. J Clin Ultrasound 2009; 37(3): 179-184. (back)
- Seward JB, Casaclang-Verzosa G. Infiltrative cardiovascular diseases: cardiomyopathies that look alike. J Am Coll Cardiol 2010; 55(17): 1769-1779. (back)
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management.J Hematol Oncol 2011; 18: 47. (back)
- Shtrasburg S, Pras M, Dolitzky M, Pariente C, Gal R, Livneh A. Pregnancy and amyloidosis: II. Suppression of amyloidogenesis during pregnancy. J Lab Clin Med 2000; 136(4): 314-319. (back)
- Siragusa S, Morice W, Gertz MA, Kyle RA, Greipp PR, Lust JA, Witzig TE, Lacy MQ, Zeldenrust SR, Rajkumar SV, Russell SJ, Hayman SR, Buadi F, Kumar SK, Dingli D, Dispenzieri A. Asymptomatic immunoglobulin light chain amyloidosis (AL) at the time of diagnostic bone marrow biopsy in newly diagnosed patients with multiple myeloma and smoldering myeloma. A series of 144 cases and a review of the literature. Ann Hematol 2011; 90(1): 101-106. (back)
- Palladini G, Dispenzieri A, Gertz MAA. Validation of the Criteria of Response to Treatment In AL Amyloidosis. Blood 2010; 116: 586-587. (back)
- Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Therneau TM. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med 1997; 336: 1202-1207. (back)
- Comenzo RL. How I treat amyloidosis. Blood 2009; 114(15): 3147-3157. (back)
- Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, Anderson JJ, O’Hara C, Finn KT, Libbey CA, Wiesman J, Quillen K, Swan N, Wright DG. High-dose melphalan andautologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med 2004; 140: 85-93. (back)
- Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin. Longterm outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood 2007; 110: 3561-3563. (back)
- Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin. Longterm outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood 2007; 110: 3561-3563. (back)
- Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Dingli D, Ansell SM, Gastineau DA, Inwards DJ, Johnston PB, Litzow MR, Micallef IN, Porrata LF, Leung N, Hogan WJ, Buadi FK. Autologous stem cell transplant for immunoglobulin light chain amyloidosis: a status report. Leuk Lymphoma 2010; 51: 2181-2187. (back)
To cite this article
Rapidly progressive renal and hepatic failure in AL-amyloidosis: bortezomib and steroid support in a young woman two months after delivery
CellR4 2014; 2 (4): e1077
Publication History
Published online: 24 Jul 2014

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.