CellR4 2014; 2 (5): e1212
Innate Immune Response and Gut Microbiota in Inflammatory Bowel Disease
Category: Reviews
Abstract
BACKGROUND: Inflammatory bowel disease (IBD) are chronic, relapsing inflammatory disorders of the digestive tract resulting from a loss of homeostasis between the intestinal immune system and the gut microbiota in genetically-predisposed individuals.
METHODS: In this review, we discuss on the emerging novel concepts on the role of innate immune response and gut microbiota in controlling mucosal homeostasis and chronic inflammation within the gastrointestinal tract.
RESULTS: Although the etiology of IBD remains largely unknown, recent evidences have focused on the interplay between innate immune response and gut microbiota modulation and have implicated the commensal microbiota as a crucial player in the unrestrained inflammatory responses observed in human and experimental IBD.
CONCLUSIONS: The information derived from these studies revealed that intestinal homeostasis and inflammation are driven by innate-type cellular elements and soluble mediators that together with gut microbiota mediate both processes, with several cytokines exhibiting opposing roles, depending upon the specific setting. This new body of information has important translational implications for both the prevention and treatment of patients suffering from IBD.
INTRODUCTION
Inflammatory bowel disease (IBD), such as Crohn’s Disease (CD) and Ulcerative Colitis (UC), are chronic, relapsing inflammatory disorders of the digestive tract resulting from a loss of homeostasis between the intestinal immune system and the gut microbiota in genetically-predisposed individuals 1. Inappropriate mucosal immune responses, due to dysregulation of tolerance to intestinal microbiota or disruption of the epithelial barrier separating microorganisms from underlying tissues, may contribute to the development or perpetuation of IBD. In fact, genome-wide association studies (GWAS) in patients with IBD have identified more than 150 associated loci, with many of the genetic variants pointing to the importance of barrier function and microbial defense 2 , 3 , 4. The new hypothesis for IBD summarizes information obtained from genetic association studies, animal models of inflammation, as well as clinical studies and observations. On the base of these data, IBD are increasingly considered a state of immunodeficiency of the innate arm of immunity, especially for a defective bacterial recognition, autophagy and antigen presentation.
In this context, the intestinal barrier represents a functional unit responsible for two main tasks that are crucial for survival of the individual: allowing nutrient absorption, and defending the body from penetration of unwanted, often dangerous, macromolecules. The gut mucosa is, in fact, a multi-layered system consisting of an external “anatomical” barrier and an inner “functional” immunological barrier. Commensal gut microbiota, the mucous layer, and the intestinal epithelial monolayer constitute the anatomical barrier. The deeper, inner layer consists of a complex network of immune cells organized in a specialized and compartmentalized system known as gut-associated lymphoid tissue or GALT. GALT represents both isolated and aggregated lymphoid follicles and is one of the largest lymphoid organs, containing up to 70% of the body’s total number of immunocytes; moreover, it is involved in response to pathogenic microorganisms and provides immune tolerance to commensal bacteria. The ability of GALT to interact with luminal antigens rests on specific mucosal immune cells (i.e., dendritic cells and M-cells), primarily localized to Peyer’s patches within the ileum that are intimately positioned at the mucosal-environmental interface and internalize microorganisms and macromolecules. These specialized immune cells have the ability to present antigen to naïve T-lymphocytes, which subsequently produce cytokines and activate mucosal immune responses, when needed. From the intracellular point of view, inflammasomes are a group of protein complexes that assemble upon recognition of a diverse set of noxious stimuli and are now considered the cornerstone of the intracellular surveillance system. Inflammasomes are able to sense both microbial and damage-associated molecular patterns (DAMPs) and initiate a potent innate, anti-microbial immune response 5. The interaction of these components sustains the maintenance of the delicate equilibrium needed for intestinal homeostasis. Many factors such as alterations in the gut microflora, modifications of the mucus layer and epithelial damage can alter this balance leading to increased intestinal permeability and translocation of luminal contents to the underlying mucosa 6. The integrity of these structures is necessary for the maintenance of normal intestinal barrier function. Dysregulation of any of the aforementioned components have been implicated not only in the pathogenesis of IBD, but many other GI disorders, including infectious enterocolitis, irritable bowel syndrome, small intestinal bowel overgrowth, and allergic food intolerance 7 , 8 , 9.
In particular, several lines of evidence have shown that the microbial flora is critical for the development of a normal gut immune system, but can also play a central role in the development of IBD 10 , 11 , 12 , 13. In support of this concept, the majority of genetically-susceptible murine models of colitis do not develop significant inflammation when raised in a germ-free environment 14 , 15 , 16 , 17, while in others, disease can be attenuated or completely abolished with antibiotic treatment 18 , 19. In this context, innate immune responses that recognize conserved microbial products, such as lipopolysaccharide (LPS) and peptidoglycan 20, are likely to be important in microbial-host interactions and intestinal homeostasis 21 21. Critical to the host’s sensing of microbes are members of the toll-like receptors (TLR) family that, alone or in combination, recognize a wide array of microbe-associated molecular patterns (MAMPs) on either pathogens or commensals 22. Furthermore, emerging evidences indicate that intestinal homeostasis and inflammation are driven by cellular elements and soluble mediators that mediate both processes, with several cytokines exhibiting opposing roles depending upon the specific setting. Related to this notion is the dogma that chronic intestinal inflammation characteristic of IBD develops through two distinct phases 23. Early disease refers to the initial events that take place when homeostatic mechanisms initially fail and acute inflammatory responses cannot be resolved. In contrast, late disease refers to the period when adaptive immunity has been irreversibly primed towards a specific effector phenotype. During these distinct stages of disease progression, innate cytokines play diverse, and often times, dichotomous roles 24 , 25.
In the present review, we will comprehensively evaluate the novel and emerging concepts about the role of innate immune response and gut microbiota in controlling mucosal homeostasis and chronic inflammation within the gastrointestinal (GI) tract. On the base of these data, we speculate about the potential implications of the modulation of these factors for treating chronic intestinal inflammation, as well as in designing more efficacious strategies for the prevention and treatment of these devastating GI pathologies.
Gut Microbiota in IBD: cause or consequence?
GI functions are carried out in a dynamic environment inhabited by 1 kg of commensal microbes that include more than 3 mln of genes 26 , 27. They belong to the three domains of life, Bacteria, Archaea and Eukarya 28 , 29 , 30, as well as to viral particles 31 , 32. Recent advances in culture-independent molecular techniques, by the analysis of phylogenetic arrays, next generation 16S rRNA sequencing and metagenome sequencing derived from human mucosal biopsies, luminal contents and feces, have shown that four major microbial phyla, (Firmicutes, Bacteroides, Proteobacteria and Actinobacteria), represent 98% of the intestinal microbiota and fall into three main groups of strict extremophile anaerobes: Bacteroides, Clostridium cluster XIVa (also known as the Clostridium Coccoides group), and Clostridium cluster IV (also known as the Clostridium leptum group) 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41 , 42.
An intricate and mutualistic symbiosis modulates the relationship between the host and the gut microbiota 43 , 44 , 45. This relationship is constantly challenged with several factors such as rapid turnover of the intestinal epithelium and overlaying mucus, exposure to peristaltic activity, food molecules, gastric, pancreatic and biliary secretions, defense molecules, drugs, pH and redox potential variations, and exposure to transient bacteria from the oral cavity and esophagus, and can lead to the collapse of the microbial community structure 46. On the other hand, resident microbes perform several useful functions, including maintaining barrier function, synthesis and metabolism of nutrients, drug and toxin metabolism, and behavioral conditioning 47. Gut microbiota is also involved in the digestion of energy substrates, production of vitamins and hormones 48, protection from pathogenic bacteria by consuming nutrients and producing molecules that inhibit their growth 49 , 50 , 51, production of nutrients for mucosal cells 52 , 53 , 54, augmenting total and pathogen-specific mucosal IgA levels upon infection 55 , 56, and in modulating immune system development and immunological tolerance 57.
Unfavorable alteration of microbiota composition, known as dysbiosis, has been implicated in chronic gut, and perhaps also systemic, immune disorders, such as in the pathogenesis of IBD, and other gastrointestinal disorders, including gastritis, peptic ulcer, irritable bowel syndrome 58 , 59 and even gastric and colon cancers 60 , 61 , 62 , 63.
Despite the growing evidence that enteric microbes may be involved in the pathogenesis of IBD in genetically susceptible individuals, there has been no convincing evidence reported to suggest that a pathogenic microorganism causes IBD. However, there is a large body of clinical and experimental data implicating the commensal microbiota as a crucial player in the inflammatory responses observed in human and experimental IBD 64. Of note, the diversion of the fecal stream from a segment of inflamed small bowel is able to reduce intestinal inflammation in patients with CD 65. On the other side, the restoration of the fecal stream to a segment of surgically resected normal bowel leaded to the induction of intestinal inflammation. This may suggest that components in the fecal stream could induce IBD 66. Moreover, dysbiosis was found in asymptomatic first-degree relatives of IBD patients, suggesting that dysbiosis may anticipate IBD 67. Several studies underlined important alterations in the composition of the microbiota in IBD patients 68. The majority of these studies found a decreased microbial diversity, especially in Firmicutes and Bacteroidetes phyla 69. Interestingly, Faecalibacterium prausnitzii, a member of the Firmicutes phyla, is significantly reduced 70 , 71 and possesses a well-documented anti-inflammatory activity 72 58. Indeed, Faecalibacterium prausnitzii is able to produce butyrate, which has a protective effect on the gut by providing energy to epithelial cells, enhancing mucosal barrier function, increasing intestinal mucous production, stimulating the production of immunosuppressive cytokines, and decreasing the generation of proinflammatory mediators, such as NF-kB 73.
On the other side, Proteobacteria and Actinobacteria are increased in patients with active IBD 74 , 75. Furthermore, specific strains of Escherichia coli are augmented in patients with CD 76. Isolates of these adherent-invasive E. coli (AIEC) have been shown to adhere to epithelial cells, and also to invade and replicate within these cells 77. Also Clostrium boltae and Clostrium symbiosum are increased in stool samples collected from patients with IBD, but even their role is not clear 78 , 79.
Despite these intriguing associations between the microbiota and IBD, however, it is not clear whether alterations in the gut microbiota represent a cause or a consequence of chronic intestinal inflammation. However, several new evidences on the IBD pathogenesis justified the substitution of the immune hyper-reactivity hypothesis with the concept of a defective innate response. In this scenario, gut microbiota acquires a leading role in IBD, which could be a disorder associated with bacterial processing. Indeed, IBD are increasingly considered a state of immunodeficiency of the innate arm of immunity with defective bacterial recognition, autophagy and antigen presentation 80. Then, the understanding that chronic intestinal inflammation develops through distinct phases and that during these separate stages of disease evolution, similar innate cellular elements and cytokines play diverse, and oftentimes, dichotomous roles has important and clear therapeutic implications.
Gut Microbiota and Innate Immune Response in IBD
In IBD, the pathological relationship between gut microbiota and innate immune response is peculiar and is crucial for triggering and exacerbating the disease. Defective epithelial barrier and increased intestinal permeability can lead to persistent immune activation and have been suggested to play a role in IBD 81. During normal gut homeostasis, small amounts of luminal antigens translocate across the epithelium both through receptor-mediated endocytosis and non-selective endocytosis. This allows the physiologic sampling of luminal content by the host’s immune system 82. Animal models that lack components of a healthy epithelial barrier have been shown to develop IBD. The lack of N-cadherin in mouse intestinal epithelium has been shown to lead to CD like symptoms 83. Moreover, alterations in the organic cation transporter (OCTN) gene, which regulates the transport of cationic proteins, such as amino acids and nutrients, are able to produce an augmented susceptibility to CD 84.
Interestingly, IBD patients have antibodies directed against several microbial antigens derived from intestinal bacteria such as E. coli and Pseudomonas fluorescens as well as yeast (e.g., Saccharomyces). In a scenario of increased gut permeability, invading microorganisms may induce intestinal immune responses that trigger the induction and the exacerbation of IBD 85. The recent identification of polymorphisms in genes that are involved in intracellular processing and killing of bacteria in patients with CD (NOD2, ATG16L1, and IRGM) suggests, indeed, that inappropriate innate immune responses to luminal bacteria could promote chronic gut inflammation in genetically susceptible individuals 86. Furthermore, the treatment with certain antibiotics is effective in reducing distal bowel inflammation in IBD patients 87.
The mucous layer that covers the intestinal epithelium represents the first physical barrier that intestinal bacteria and food antigens encounter on the mucosal surface. It provides protection by shielding the epithelium from microorganisms and harmful antigens, while acting as a lubrificant for intestinal motility. It consists of two layers: an inner layer and an outer layer. These mucus layers are organized around the highly glycosylated MUC2 mucin, forming a large, net-like polymer that is secreted by the goblet cells. The inner mucus layer is dense and does not allow bacteria to penetrate, thus keeping the epithelial cell surface free from bacteria. The inner mucus layer is converted into the outer layer, which is the habitat of the commensal flora. The outer mucus layer has an expanded volume due to the proteolytic activities provided by the host but probably also caused by commensal bacterial proteases and glycosidases 88. This compartmentalization seems to be fundamental for the homeostasis in the highly colonized colon. The importance of the mucus barrier was further demonstrated in Muc2 deficient mice where bacteria are in direct contact with the epithelial cells and are also found deep in the crypts as well as inside epithelial cells 89. Loss of the barrier formed by the inner mucus layer triggers spontaneous colitis and development of colon cancer 90 , 91 , 92. Moreover, some CD patients have goblet cell depletion and an impaired mucus layer, which allows bacteria to adhere directly to epithelial cells, and this may contribute to disease progression 93.
The second line of defense against bacteria invasion is formed by the intestinal epithelium, which consists of enterocytes and specialized epithelial cells, such as goblet cells and Paneth cells. Besides the formation of a physical barrier against bacteria, epithelial cells can secrete a number of antimicrobial peptides. Defective expression of antimicrobial peptides has been observed in patients with CD 94. Paneth cells are a further intestinal epithelial cell 95 type that plays an important role in mucosal homeostasis, and, if functionally impaired, may contribute to IBD 96. Paneth cells reside at the base of small intestinal crypts and secrete AMPs as well as inflammatory mediators 97.
The epithelium lies between the immune cells in the lamina propria and the microbiota in the gut lumen and it functions to communicate with both. The microbiota signals enterocytes as well as innate cells in the lamina propria via pattern recognition molecules signal receptors, such as cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and TLRs. These signals have been shown to be necessary for normal homeostasis and resistance to injury 98.
NOD2 is a protein that acts as an intracellular pattern recognition receptor for muramyl dipeptide (MDP), a component of the bacterial wall peptidoglycans. Deficient mice for intracellular pattern recognition receptors, NOD1 and NOD2, had decreased E-cadherin expression with increased epithelial permeability and decreased antimicrobial production 99. Polymorphisms in the card15/nod2 gene have been constantly associated with increased risk for developing CD 100. Notably, patients harboring NOD2 risk variants express decreased levels of human α-defensins 5 and 6 (HD5, HD6) in Paneth cells 101. Although the functional role of NOD2 mutations is still controversial, available evidence suggests that they represent loss-of- function mutations that lead to a reduced activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) 102. Furthermore, Marks et al demonstrated that CD patients may also carry a primary defect in phagocytic function 103.Interestingly, he found that this defect could be independent of the CARD15 polymorphism status, indicating that patients with an intact genotype may still have defective NOD2 signaling. Recent studies in SAMP1/YitFc mice with CD-like ileitis (but intact CARD15 genotype) demonstrate defective MDP-mediated responses, including innate cytokine secretion 104, and give further support to this hypothesis.
Analyzing all the gene mutations associated to IBD, the majority of them fall into few distinct pathophysiological categories, thereby pointing to a limited number of inherent defects 105. The main part of these abnormalities clearly relate to the function of the innate immune system and effective recognition, intracellular manipulation, and elimination of bacterial factors. These findings strongly support the concept of impaired innate immunity in IBD, thereby leading to defective microbial clearance and persistent antigenic stimulation.
TLRs are expressed on cells within the gut mucosa, including IECs and more broadly, on lamina propria macrophages and dendritic cells 106 , 107 , 108 , 109. TLR2, -4 and -5 are the major cell-surface sensors of bacterial lipopeptides, including LPS and flagellins, while TLR3, -7, -8 and -9 detect nucleic acid motifs 110. Among the described TLRs, both human and murine studies have shown the importance of TLR5 and its ligand, the bacterial protein flagellin, which is the major structural component of bacterial flagella, in the regulation of innate and adaptive immune responses that are associated with IBD 111 , 112 , 113 , 114 . However, results from the currently published data do not lead to a definitive conclusion regarding the precise role of TLR5, but open the possibilities to different mechanistic hypotheses. Commensal-derived flagellin has been identified as a dominant antigen in patients with CD, and about 50% of CD patients usually have abnormally high levels of anti-flagellin antibodies that correlate with particular subtypes of severe disease 115 , 116. Furthermore, a TLR5 stop codon polymorphism, which prevents TLR5 signaling, reduces anti-flagellin adaptive immune responses and appears to be protective against CD in certain ethnic groups 117. Conversely, some studies have documented a downregulation of TLR5 in human IBD 118 , 119. In mice, TLR5-deficient animals develop spontaneous colitis in some animal facilities 120 and are more susceptible to dextran sulfate sodium (DSS)-induced colitis, a T-cell independent, chemically-induced model of epithelial damage and acute inflammation, primarily driven by innate immune responses 121. In contrast, flagellin resulted increased in T. gondii-induced ileitis 122. As such, the role of flagellin/TLR5 in the pathogenesis of IBD is controversial (i.e., pathogenic vs. protective) and the cell types that express TLR5, as well as the TLR5-dependent mechanisms that modulate gut permeability and homeostasis towards gut microbiota remain unclear.
The dichotomous role of Toll/IL-1 Receptor superfamily in IBD
The behavior of the cells mediating innate immunity is altered significantly in individuals with IBD. These cells are orchestrated by specific cytokines, such as those of the interleukin (IL)-1 family. The role of the Toll/IL-1 Receptor (TIR) superfamily and their respective ligands, of which IL-1-like molecules belong, is well established in the pathogenesis of several autoinflammatory and chronic immune disorders 123. However, the emerging concept that TLRs, as well as IL-1 and its related cytokine family members, also play a critical role in health and maintenance of immune homeostasis is gaining increasing acceptance 124. The GI system, in fact, represents one of the best examples of where these opposing mechanisms simultaneously take place 125. A large body of evidence exists that support the contribution of various IL-1 family members, particularly IL-1 and IL-18, to the pathogenesis of IBD, as well as GI-related cancers. However, while selective blockade of proinflammatory cytokines is one of the most effective strategies to down-regulate mucosal inflammation in IBD 126, Phase I clinical trials using strategies to neutralize either IL-1 or IL-18 have failed to show significant efficacy in treating patients with UC and CD, respectively. One potential cause for this failure is the dichotomous functions of these IL-1 family members in inducing disease pathogenesis, while simultaneously promoting protection, within the intestinal gut mucosa.
New insights into the role of cytokine-driven pathways in mucosal immunity have been described, based on several recent studies in animal models of acute intestinal injury, repair, and chronic inflammation. Information derived from these studies reveal that intestinal homeostasis and inflammation are driven by cellular elements and soluble mediators that mediate both processes, with several cytokines exhibiting opposing roles, depending upon the specific setting. This concept is most strongly supported by members of the IL-1 family of cytokines in the pathogenesis of IBD 127 , 128 , 129 , 130 , 131 , 132 , 133 , 134 , 135 , 136 , 137 , 138 , 139 , 140, where the same cytokine can possess both classic pro-inflammatory properties, as well as protective, anti-inflammatory functions, which is primarily dependent on the presence of receptor-bearing cells during the host’s disease state.
As such, aside from the established pro-inflammatory properties of IL-1α, IL-1β, IL-18 and their downstream signaling molecules shared with TLR family members, such as NF-kB and myeloid differentiation primary response 88 (MyD88), a growing body of evidence indicates that these mediators are necessary for the maintenance of mucosal homeostasis by effectively handling microbiota, as well as by protecting and restoring the integrity of the epithelial barrier 141 , 142 , 143. While little is known regarding the potential contributions of other IL-1 family members, such as IL-36, IL-36Ra, IL-37, and IL-38, in chronic intestinal inflammation and gut health, the evolving literature regarding the role of IL-33, the most recently described IL-1 family member is, at present, ambiguous and may reflect yet another example of an innate-type cytokine that possesses multiple functions depending on the immunological status and genetic susceptibility of the host. Although one of the first observations of IL-33-dependent functions in the gut was potent epithelial proliferation and mucus production 144, suggesting the promotion of mucosal repair and healing, dysregulated or uncontrolled IL-33 production may also lead to more pathogenic features characteristic of IBD, including epithelial barrier dysfunction, chronic, relapsing inflammation, and formation of fibrotic lesions 145 , 146. In general, early activation of the intestinal epithelium by pathogenic organisms and/or other noxious environmental antigens elicits the production of epithelial-derived IL-1 family members, including intracellular (ic)IL-1Ra, IL-1α, IL-18, and IL-33. Epithelial disruption often occurs, facilitating translocation of luminal bacterial products and the recruitment of innate immune cells, primarily neutrophils and macrophages that are also a potent source of secreted (s)IL-1Ra, IL-1β, IL-18, and IL-33. Normally, early expression of these mediators dampen acute inflammation and promote epithelial repair and restitution, with the ultimate goal of limiting gut mucosal damage and restoring intestinal homeostasis. Under conditions of either uncontrolled and/or persistent inflammation (e.g., as a result of innate immune dysfunction or host genetic predisposition), infiltration of adaptive immune cells, bearing various IL-1R family members, occurs during the later phases of inflammation, making available an effector population able to respond to IL-1-like ligands. For example, the presence of naïve CD4+ T cells expressing the IL-18R have the ability to respond to IL-18, and in combination with IL-12, represents one of the most potent stimuli for interferon (IFN)γ production and Th1 polarized effector responses, thereby promoting chronic Th1-mediated inflammation. Similar effects can occur upon IL-33 stimulation of naïve CD4+ T cells, but in this case, a robust Th2 immune response results. Furthermore, several levels of regulation exist within each subfamily of IL-1 family members, often including the presence of several agonist isoforms (both precursor and mature, cleaved forms), receptor antagonists, as well as soluble and cell-bound decoy receptors. In addition, the promiscuity of IL-1 family ligands with both binding receptors as well as recruited accessory proteins, instills yet another level of regulation that should be considered when determining the overall biological effects of a specific IL-1 family member agonist. In fact, IL-1 family members cannot be considered in isolation, but with other IL-1-related proteins that can influence their overall interactive effects. An imbalance in the equilibrium between IL-1 family components, dependent on prevalent isoform and receptor binding domain/accessory protein present on effector cells, may be responsible for either driving pathogenic events, including chronic intestinal inflammation, fibrosis, and CRC, or for promoting protection by inducing epithelial repair, mucosal wound healing, and restoration of gut homeostasis (summarized in Figure 1). Based on this new information and the emerging concept that IL-1 family members can possess opposing role in gut health and disease, novel pathogenic hypotheses can be formed with important translational implications in regard to the prevention and treatment of chronic intestinal inflammation, including CD and UC, and CRC.
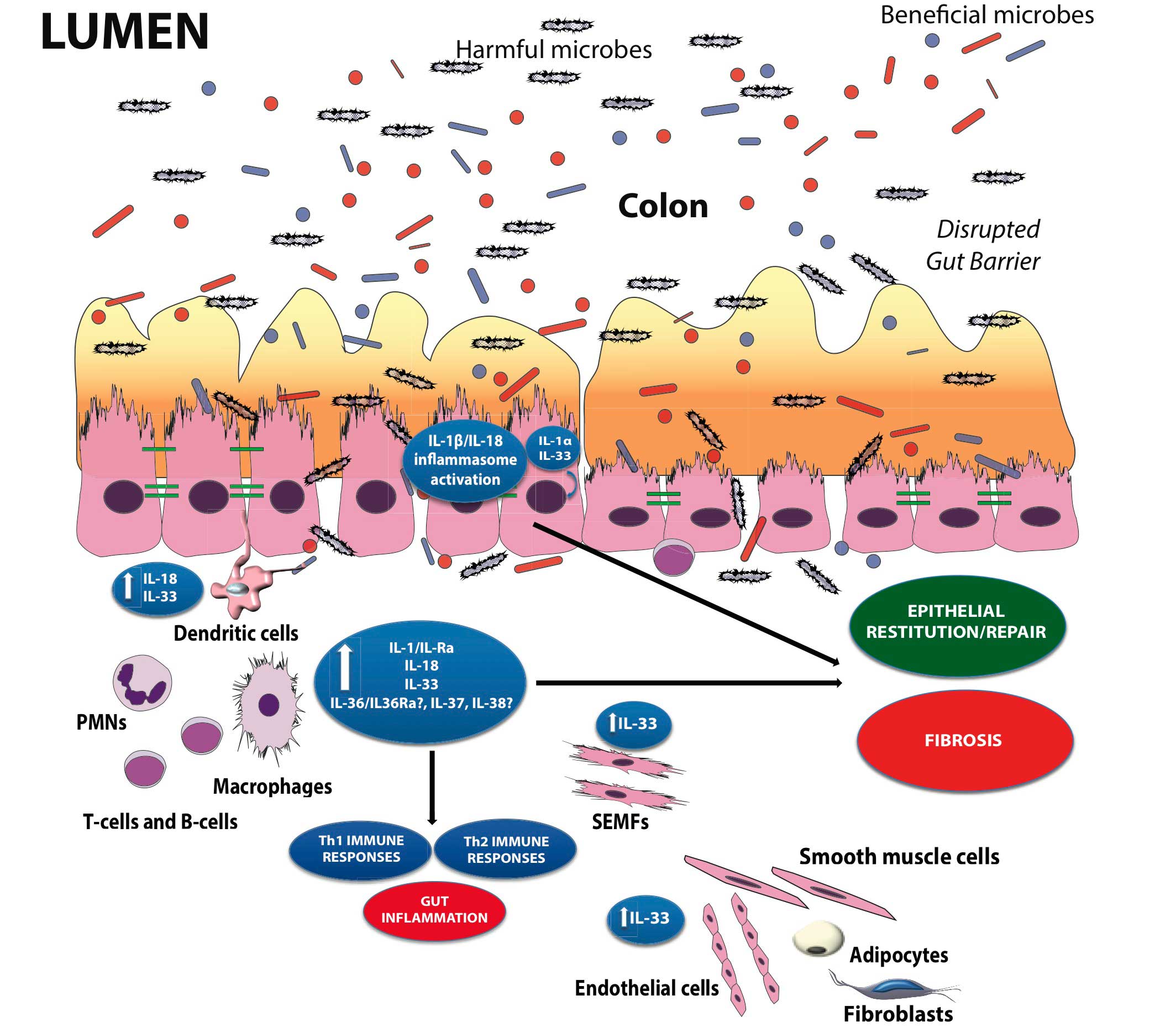
Figure 1. The dichotomous role of IL-1 family members within the gut mucosa. The balance between proinflammatory and protective cytokines is crucial for the maintenance of gut homeostasis. Damage to the epithelium and other proinflammatory stimuli, including PAMPs derived from luminal antigens and the local intestinal microflora, induce the expression of IL-1 family members that are subsequently released by necrotic IECs as potential alarmins (e.g., IL-33 and IL-1α). Depending on the cellular source and presence of receptor-bearing effector cells, IL-1 family members can possess very different and often opposite functions within the gut mucosa. Therapeutic interventions should consider all this processes and whether targeting specific IL-1 family members may be more efficacious during active disease vs. maintaining remission.
CONCLUSIONS
The present review provides an analysis on the interplay between gut microbiota and innate immune response in gut chronic inflammation. In IBD, the pathological relationship between gut microbiota and innate immune response is peculiar and is crucial for triggering and exacerbating the disease. Information derived from several studies reveal that intestinal homeostasis and inflammation are driven by cellular elements and innate-type soluble mediators that mediate both processes, with several cytokines exhibiting opposing roles, depending upon the specific setting and disease phase. This intriguingly concept has suggested that IBD are strictly correlated to defective innate response and not to a mucosal immune hyper-reactivity. Related to this notion is the dogma that chronic intestinal inflammation characteristic of IBD develops through an early and late disease. It will be fascinating to completely elucidate the underlying mechanisms for this new concept in order to provide indications for unexplained patient-to-patient variations in drug efficacy and toxicity. In this scenario, the goal of early phase treatment should promote innate immune response, while late disease will continue to use anti-inflammatory drugs. It will also be important to perform detailed mechanistic studies to improve the development of microbial therapies that may modulate the composition of the gut microflora, with the end goal of promoting gut health.
Conflict of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59(3): 451-459. (back)
- Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448(7152): 427-434. (back)
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491(7422): 119-124. (back)
- Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011; 474(7351): 307-317. (back)
- Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature 2012; 481(7381): 278-286. (back)
- Scaldaferri F, Pizzoferrato M, Gerardi V, Lopetuso L, Gasbarrini A. The gut barrier: new acquisitions and therapeutic approaches. J Clin Gastroenterol 2012;4 6 Suppl: S12-17. (back)
- Camilleri M, Madsen K, Spiller R, Van Meerveld BG, Verne GN. Intestinal barrier function in health and gastrointestinal disease. Neurogastroenterol Motil 2012; 24(6): 503-512. (back)
- Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol 2012; 42(1): 71-78. (back)
- Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev 2011; 91(1): 151-175. (back)
- Peloquin JM, Nguyen DD. The microbiota and inflammatory bowel disease: Insights from animal models. Anaerobe 2013; 24: 102-106. (back)
- Scaldaferri F, Petito V, Lopetuso L, Bruno G, Gerardi V, Ianiro G, et al. Pre- and posttherapy assessment of intestinal soluble mediators in IBD: where we stand and future perspectives. Mediators Inflamm 2013; 2013: 391473. (back)
- Scaldaferri F, Gerardi V, Lopetuso LR, Del Zompo F, Mangiola F, Boskoski I, et al. Gut microbial flora, prebiotics, and probiotics in IBD: their current usage and utility. Biomed Res Int 2013; 2013: 435268. (back)
- Purchiaroni F, Tortora A, Gabrielli M, Bertucci F, Gigante G, Ianiro G, et al. The role of intestinal microbiota and the immune system. Eur Rev Med Pharmacol Sci 2013; 17(3): 323-333. (back)
- Mombaerts P, Mizoguchi E, Grusby MJ, Glimcher LH, Bhan AK, Tonegawa S. Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 1993; 75(2): 274-282. (back)
- Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993; 75(2): 253-261. (back)
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993; 75(2): 263-274. (back)
- Blumberg R, Powrie F. Microbiota, disease, and back to health: a metastable journey. Sci Transl Med 2012; 4(137): 137rv7. (back)
- Madsen KL, Doyle JS, Tavernini MM, Jewell LD, Rennie RP, Fedorak RN. Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology 2000; 118(6): 1094-1105. (back)
- Kang SS, Bloom SM, Norian LA, Geske MJ, Flavell RA, Stappenbeck TS, et al. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med 2008; 5(3): e41. (back)
- Guarino A, Albano F, Ashkenazi S, Gendrel D, Hoekstra JH, Shamir R, et al. European Society for Paediatric Gastroenterology, Hepatology, and Nutrition/European Society for Paediatric Infectious Diseases evidence-based guidelines for the management of acute gastroenteritis in children in Europe: executive summary. J Pediatr Gastroenterol Nutr 2008; 46(5): 619-621. (back)
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124(4): 783-801. (back)
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124(4): 783-801. (back)
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59(3): 451-459. (back)
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59(3): 451-459. (back)
- Lopetuso LR, Chowdhry S, Pizarro TT. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Front Immunol 2013; 4: 181. (back)
- Leser TD, Molbak L. Better living through microbial action: the benefits of the mammalian gastrointestinal microbiota on the host. Environ Microbiol 2009; 11(9): 2194-2206. (back)
- Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology 2009; 136(1): 65-80. (back)
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005; 308(5728): 1635-1638. (back)
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science 2006; 312(5778): 1355-1359. (back)
- Scanlan PD, Marchesi JR. Micro-eukaryotic diversity of the human distal gut microbiota: qualitative assessment using culture-dependent and -independent analysis of faeces. ISME J 2008; 2(12): 1183-1193. (back)
- Zhang T, Breitbart M, Lee WH, Run JQ, Wei CL, Soh SW, et al. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol 2006; 4(1): e3. (back)
- Breitbart M, Haynes M, Kelley S, Angly F, Edwards RA, Felts B, et al. Viral diversity and dynamics in an infant gut. Res Microbiol 2008; 159(5): 367-373. (back)
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005; 308(5728): 1635-1638. (back)
- Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science 2006; 312(5778): 1355-1359. (back)
- Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol 2002; 39(1): 33-39. (back)
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005; 307(5717): 1915-1920. (back)
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005; 102(31): 11070-11075. (back)
- Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006; 124(4): 837-848. (back)
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104(34): 13780-13785. (back)
- Rajilic-Stojanovic M, Smidt H, de Vos WM. Diversity of the human gastrointestinal tract microbiota revisited. Environ Microbiol 2007; 9(9): 2125-2136. (back)
- Tap J, Mondot S, Levenez F, Pelletier E, Caron C, Furet JP, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol 2009; 11(10): 2574-2584. (back)
- Manson JM, Rauch M, Gilmore MS. The commensal microbiology of the gastrointestinal tract. Adv Exp Med Biol 2008; 635: 15-28. (back)
- Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005; 307(5717): 1915-1920. (back)
- McCracken VJ, Lorenz RG. The gastrointestinal ecosystem: a precarious alliance among epithelium, immunity and microbiota. Cell Microbiol 2001; 3(1): 1-11. (back)
- Lievin-Le Moal V, Servin AL. The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev 2006; 19(2): 315-337. (back)
- Manson JM, Rauch M, Gilmore MS. The commensal microbiology of the gastrointestinal tract. Adv Exp Med Biol 2008; 635: 15-28. (back)
- Scaldaferri F, Pizzoferrato M, Gerardi V, Lopetuso L, Gasbarrini A. The gut barrier: new acquisitions and therapeutic approaches. J Clin Gastroenterol 2012;4 6 Suppl: S12-17. (back)
- Sekirov I. RS, Antunes LC, Finlay BB. Gut mircobiota in health and disease. Physiol Rev 2010; 90: 859-904. (back)
- Silva AM BF, Duarte R, Vieira LQ, Arantes RM, Nicoli JR. Effect of Bifidobacterium longum ingestion on experimental salmonellosis in mice. J Appl Microbiol 2004; 97: 29-37. (back)
- Truusalu K MRea. Eradication of Salmonella Typhimurium infection in a murine model of typhoid fever with the cimbination of probiotic Lactobacillus fermentum ME-3 and ofloxacin. BMC Microbiol 2008; 8: 132. (back)
- Searle LE, Best A, Nunez A, Salguero FJ, Johnson L, Weyer U, et al. A mixture containing galactooligosaccharide, produced by the enzymic activity of Bifidobacterium bifidum, reduces Salmonella enterica serovar Typhimurium infection in mice. J Med Microbiol 2009; 58(Pt 1): 37-48. (back)
- Martens EC RRea. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent gut symbiont. J Biol Chem 2009; 284: 18445-18457. (back)
- Burger van Paassen N VAea. The regulation of intestinal mucin MUC2 expression by short chain fatty acid: implications for epithelial pretection. Biochem J 2009; 420: 211-219. (back)
- Dharmani P, Srivastava V, Kissoon-Singh V, Chadee K. Role of intestinal mucins in annate host defense mechanisms against pathogens. J Innamte Immun 2009; 1: 123-135. (back)
- Galdeano CM, Perdigon G. The probiotic bacterium Lactobacillus casei induces activation of the gut mucosal immune system through innate immunity. Clin Vaccine Immunol 2006; 13(2): 219-226. (back)
- Leblanc J, Fliss I, Matar C. Induction of a humoral immune response following an Escherichia coli O157:H7 infection with an immunomodulatory peptidic fraction derived from Lactobacillus helveticus-fermented milk. Clin Diagn Lab Immunol 2004; 11(6): 1171-1181. (back)
- Allen CA, Torres AG. Host-microbe communication within the GI tract. Adv Exp Med Biol 2008; 635: 93-101. (back)
- Parashar UD, Gibson CJ, Bresee JS, Glass RI. Rotavirus and severe childhood diarrhea. Emerg Infect Dis 2006; 12(2): 304-306. (back)
- Scaldaferri F, Nardone O, Lopetuso LR, Petito V, Bibbo S, Laterza L, Gerardi V, Bruno G, Scoleri I, Diroma A, Sgambato A, Gaetani E, Cammarota G, Gasbarrini A. Intestinal gas production and gastrointestinal symptoms: from pathogenesis to clinical implication. Eur Rev Med Pharmacol Sci 2013; 17 Suppl 2: 2-10. (back)
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104(34): 13780-13785. (back)
- Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002; 122(1): 44-54. (back)
- Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol 2010; 28: 623-667. (back)
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008; 134(2): 577-594. (back)
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008; 134(2): 577-594. (back)
- Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R, et al. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991; 338(8770): 771-774. (back)
- Rutgeerts P, Goboes K, Peeters M, Hiele M, Penninckx F, Aerts R, et al. Effect of faecal stream diversion on recurrence of Crohn’s disease in the neoterminal ileum. Lancet 1991; 338(8770): 771-774. (back)
- Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011; 60(5): 631-637. (back)
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008; 134(2): 577-594. (back)
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008; 134(2): 577-594. (back)
- Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011; 140(6): 1720-1728. (back)
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105(43): 16731-16736. (back)
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105(43): 16731-16736. (back)
- Looijer-van Langen MA, Dieleman LA. Prebiotics in chronic intestinal inflammation. Inflamm Bowel Dis 2009; 15(3): 454-462. (back)
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008; 134(2): 577-594. (back)
- Koboziev I, Reinoso Webb C, Furr KL, Grisham MB. Role of the enteric microbiota in intestinal homeostasis and inflammation. Free Radic Biol Med 2013; 68C: 122-133. (back)
- Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011; 140(6): 1720-1728. (back)
- Chassaing B, Rolhion N, de Vallee A, Salim SY, Prorok-Hamon M, Neut C, et al. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J Clin Invest 2011; 121(3): 966-975. (back)
- Lozupone C, Faust K, Raes J, Faith JJ, Frank DN, Zaneveld J, et al. Identifying genomic and metabolic features that can underlie early successional and opportunistic lifestyles of human gut symbionts. Genome Res 2012; 22(10): 1974-1984. (back)
- Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A. Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 2013; 5(1): 23. (back)
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59(3): 451-459. (back)
- Salim SY, Soderholm JD. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm Bowel Dis 2011; 17(1): 362-381. (back)
- Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 2009; 325(5940): 617-620. (back)
- Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 1995; 270(5239): 1203-1207. (back)
- Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet 2004; 36(5): 471-475. (back)
- 57 (back)
- Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011; 140(6): 1720-1728. (back)
- Feller M, Huwiler K, Schoepfer A, Shang A, Furrer H, Egger M. Long-term antibiotic treatment for Crohn’s disease: systematic review and meta-analysis of placebo-controlled trials. Clin Infect Dis 2010; 50(4): 473-480. (back)
- Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A 2010; 108 Suppl 1: 4659-4665. (back)
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 2008; 105(39): 15064-15069. (back)
- Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 2008; 105(39): 15064-15069. (back)
- Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002; 295(5560): 1726-1729. (back)
- Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006; 131(1): 117-129. (back)
- Larsson JM, Karlsson H, Crespo JG, Johansson ME, Eklund L, Sjovall H, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis 2011; 17(11): 2299-2307. (back)
- Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2003; 9(4): 215-223. (back)
- Stanislawowski M, Wierzbicki PM, Golab A, Adrych K, Kartanowicz D, Wypych J, et al. Decreased Toll-like receptor-5 (TLR-5) expression in the mucosa of ulcerative colitis patients. J Physiol Pharmacol 2009; 60 Suppl 4: 71-75. (back)
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010; 28: 573-621. (back)
- Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 2011; 9(5): 356-368. (back)
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004; 118(2): 229-241. (back)
- Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, et al. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1-/-; Nod2-/- mice. Inflamm Bowel Dis 2012; 18(8): 1434-1446. (back)
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001; 411(6837): 599-603. (back)
- Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci U S A 2005; 102(50): 18129-18134. (back)
- Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 2004; 53(11): 1658-1664. (back)
- Marks DJ, Harbord MW, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, et al. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet 2006; 367(9511): 668-678. (back)
- Corridoni D, Kodani T, Rodriguez-Palacios A, Pizarro TT, Xin W, Nickerson KP, et al. Dysregulated NOD2 predisposes SAMP1/YitFc mice to chronic intestinal inflammation. Proc Natl Acad Sci U S A 2013; 110(42): 16999-17004. (back)
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59(3): 451-459. (back)
- Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 2000; 68(12): 7010-7017. (back)
- Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 2001; 167(4): 1882-1885. (back)
- Abreu MT, Arnold ET, Thomas LS, Gonsky R, Zhou Y, Hu B, et al. TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J Biol Chem 2002; 277(23): 20431-20437. (back)
- Hershberg RM. The epithelial cell cytoskeleton and intracellular trafficking. V. Polarized compartmentalization of antigen processing and Toll-like receptor signaling in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 2002; 283(4): G833-839. (back)
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000; 406(6797): 782-787. (back)
- Peloquin JM, Nguyen DD. The microbiota and inflammatory bowel disease: insights from animal models. Anaerobe 2013; 24: 102-106. (back)
- Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, et al. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest 2004; 113(9): 1296-1306. (back)
- Dubinsky MC, Kugathasan S, Mei L, Picornell Y, Nebel J, Wrobel I, et al. Increased immune reactivity predicts aggressive complicating Crohn’s disease in children. Clin Gastroenterol Hepatol 2008; 6(10): 1105-1111. (back)
- Targan SR, Landers CJ, Yang H, Lodes MJ, Cong Y, Papadakis KA, et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology 2005; 128(7): 2020-2028. (back)
- Dubinsky MC, Kugathasan S, Mei L, Picornell Y, Nebel J, Wrobel I, et al. Increased immune reactivity predicts aggressive complicating Crohn’s disease in children. Clin Gastroenterol Hepatol 2008; 6(10): 1105-1111. (back)
- Targan SR, Landers CJ, Yang H, Lodes MJ, Cong Y, Papadakis KA, et al. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn’s disease. Gastroenterology 2005; 128(7): 2020-2028. (back)
- Gewirtz AT, Vijay-Kumar M, Brant SR, Duerr RH, Nicolae DL, Cho JH. Dominant-negative TLR5 polymorphism reduces adaptive immune response to flagellin and negatively associates with Crohn’s disease. Am J Physiol Gastrointest Liver Physiol 2006; 290(6): G1157-1163. (back)
- Stanislawowski M, Wierzbicki PM, Golab A, Adrych K, Kartanowicz D, Wypych J, et al. Decreased Toll-like receptor-5 (TLR-5) expression in the mucosa of ulcerative colitis patients. J Physiol Pharmacol 2009; 60 Suppl 4: 71-75. (back)
- Ortega-Cava CF, Ishihara S, Rumi MA, Aziz MM, Kazumori H, Yuki T, et al. Epithelial toll-like receptor 5 is constitutively localized in the mouse cecum and exhibits distinctive down-regulation during experimental colitis. Clin Vaccine Immunol 2006; 13(1): 132-138. (back)
- Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest 2007; 117(12): 3909-3921. (back)
- Ivison SM, Himmel ME, Hardenberg G, Wark PA, Kifayet A, Levings MK, et al. TLR5 is not required for flagellin-mediated exacerbation of DSS colitis. Inflamm Bowel Dis 2010; 16(3): 401-409. (back)
- Erridge C, Duncan SH, Bereswill S, Heimesaat MM. The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PLoS One 2010; 5(2): e9125. (back)
- Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011; 117(14): 3720-3732. (back)
- Lopetuso LR, Chowdhry S, Pizarro TT. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Front Immunol 2013; 4: 181. (back)
- Pizarro TT, Cominelli F. Cytokine therapy for Crohn’s disease: advances in translational research. Annu Rev Med 2007; 58: 433-444. (back)
- Rutgeerts P, Vermeire S, Van Assche G. Biological therapies for inflammatory bowel diseases. Gastroenterology 2009; 136(4): 1182-1197. (back)
- Cominelli F, Nast CC, Clark BD, Schindler R, Lierena R, Eysselein VE, et al. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J Clin Invest 1990; 86(3): 972-980. (back)
- Andus T, Daig R, Vogl D, Aschenbrenner E, Lock G, Hollerbach S, et al. Imbalance of the interleukin 1 system in colonic mucosa–association with intestinal inflammation and interleukin 1 receptor antagonist [corrected] genotype 2. Gut 1997; 41(5): 651-657. (back)
- Nishiyama T, Mitsuyama K, Toyonaga A, Sasaki E, Tanikawa K. Colonic mucosal interleukin 1 receptor antagonist in inflammatory bowel disease. Digestion 1994; 55(6): 368-373. (back)
- Ferretti M, Casini-Raggi V, Pizarro TT, Eisenberg SP, Nast CC, Cominelli F. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest 1994; 94(1): 449-453. (back)
- Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. J Immunol 1995; 154(5): 2434-2440. (back)
- Pizarro TT, Michie MH, Bentz M, Woraratanadharm J, Smith MF, Jr., Foley E, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol 1999; 162(11): 6829-6835. (back)
- Monteleone G, Trapasso F, Parrello T, Biancone L, Stella A, Iuliano R, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol 1999; 163(1): 143-147. (back)
- McNamee EN, Masterson JC, Jedlicka P, McManus M, Grenz A, Collins CB, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci U S A 2011; 108(40): 16711-16716. (back)
- Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A 2010; 107(17): 8017-8022. (back)
- Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis 2010; 16(7): 1097-1107. (back)
- Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol 2010; 45(10): 999-1007. (back)
- Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett 2010; 128(1): 80-85. (back)
- Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine 2012; 59: 451-459. (back)
- Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarstrom C, Loos T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol 2010; 177(6): 2804-2815. (back)
- Kojouharoff G, Hans W, Obermeier F, Mannel DN, Andus T, Scholmerich J, et al. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin Exp Immunol 1997; 107(2): 353-358. (back)
- Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med 2002; 8(10): 1089-1097. (back)
- Reuter BK, Pizarro TT. Commentary: the role of the IL-18 system and other members of the IL-1R/TLR superfamily in innate mucosal immunity and the pathogenesis of inflammatory bowel disease: friend or foe? Eur J Immunol 2004; 34(9): 2347-2355. (back)
- Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005; 23(5): 479-490. (back)
- Lopetuso LR, Scaldaferri F, Pizarro TT. Emerging role of the interleukin (IL)-33/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair 2012; 5(1): 18. (back)
- Pastorelli L, De Salvo C, Cominelli MA, Vecchi M, Pizarro TT. Novel cytokine signaling pathways in inflammatory bowel disease: insight into the dichotomous functions of IL-33 during chronic intestinal inflammation. Therap Adv Gastroenterol 2011; 4(5): 311-323. (back)
To cite this article
Innate Immune Response and Gut Microbiota in Inflammatory Bowel Disease
CellR4 2014; 2 (5): e1212
Publication History
Published online: 30 Sep 2014

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.